
 Tiếng Việt
Tiếng Việt 日本語
日本語Selecting a medical device EMS provider is not a procurement decision — it is a regulatory decision. The contract manufacturer you choose becomes an extension of your quality system. Their processes appear in your Device History Records. Their change control failures become your FDA warning letters. Their traceability gaps become your recall exposure. In medical electronics manufacturing, the line between your quality system and your EMS partner’s quality system is not a boundary — it is a shared responsibility. Medical OEMs look to their medical device EMS provider to offer not only the best processes and highest quality standards, but the industry expertise and experience that can guide new product introductions seamlessly through development — from first prototype build through FDA submission and into sustained mass production. For U.S. and EU OEMs evaluating contract manufacturing options in 2026, Vietnam has emerged as a strategically compelling location for medical device electronics manufacturing. The combination of ISO 13485-certified EMS capability, significantly lower tariff exposure versus China, and a growing engineering talent base makes Vietnam the most viable Asia-Pacific alternative for medical-grade electronics production.
This guide covers what a qualified medical device EMS provider must demonstrate — ISO 13485 certification, full component traceability, FDA-ready process validation, change control, and CAPA systems — and what U.S. OEMs should verify before awarding a medical device program to any contract manufacturer, anywhere in the world.
What Makes an EMS Provider “Medical-Grade”?
Not every EMS provider that claims medical device capability actually has it. The gap between a commercial EMS running IPC Class 2 processes and a genuine medical device EMS provider operating under ISO 13485 is wide — and the consequences of choosing the wrong partner are measured in FDA citations, product recalls, and patient safety events.
Medical Electronics Manufacturing vs. Standard Commercial EMS

The fundamental difference between commercial and medical electronics manufacturing is not equipment — it is discipline. A commercial EMS optimizes for cost and throughput. A medical device EMS provider optimizes for process consistency, traceability, and regulatory defensibility.
Medical devices operate in highly regulated environments where failure is not an option. Unlike commercial products, medical electronics require strict process validation, full traceability, controlled manufacturing environments, extensive documentation, and long product lifecycle support. Even small manufacturing inconsistencies can create significant regulatory and operational risks.
The five requirements that distinguish a genuine medical device EMS provider from a commercial EMS claiming medical capability:
- Documented Quality Management System (QMS) aligned to ISO 13485 — not ISO 9001, not “equivalent,” but ISO 13485 with a scope statement that explicitly covers electronics assembly for medical devices
- Full component-level traceability — lot number, date code, and supplier Certificate of Conformance (CoC) for every component on every board, linked to a specific unit serial number
- Process validation — IQ/OQ/PQ documentation for every critical manufacturing process, demonstrating that the process consistently produces product meeting specification
- Formal change control — every change to component, process, or supplier must be documented, impact-assessed, and approved before implementation
- Device History Record (DHR) — a complete, legally defensible build record for every unit produced, maintained in a format that supports FDA audit and recall response
| Requirement | Commercial EMS | Medical Device EMS Provider |
|---|---|---|
| Quality system | ISO 9001 (optional) | ISO 13485 (mandatory) |
| Traceability | Board-level | Component lot + serial number |
| Process validation | Not required | IQ/OQ/PQ documented |
| Change control | Informal | Formal ECO with OEM approval |
| Build records | Production log | Full DHR per unit |
| Supplier qualification | Price-driven | Approved Supplier List (ASL) |
| Inspection standard | IPC Class 2 | IPC Class 3 |
IPC Class 3: The Manufacturing Standard for Medical Electronics
A medical device EMS provider must build to IPC Class 3 — the highest reliability level defined in IPC standards, designed for electronics where continued performance is critical and equipment downtime cannot be tolerated.
IPC Class 3 imposes tighter acceptance criteria at every inspection point: solder joint geometry, component placement accuracy, minimum annular ring dimensions, and solder mask integrity. 100% inspection is required — statistical sampling is not acceptable for Class 3 assemblies.
Key differences between IPC Class 2 and Class 3 in production practice:
- Solder joint acceptance: Class 3 requires minimum 75% solder fill in through-hole barrels vs. 50% for Class 2
- Component placement: tighter offset tolerances — components must be more precisely centered on pads
- AOI and X-ray: mandatory for all hidden solder joints (BGA, QFN, LGA) — not optional
- Operator certification: IPC-A-610 Certified Inspectors required; J-STD-001 certified operators for soldering
IPC-A-610 Certified Trainers and Certified Product Specialists who understand the unique acceptance criteria for medical applications ensure workmanship meets or exceeds Class 3 requirements throughout the production lifecycle.
FDA Regulatory Framework: What EMS Providers Need to Understand
FDA 21 CFR Part 820 (Quality System Regulation) applies not only to medical device OEMs but to their contract manufacturers. A medical device EMS provider that does not understand its obligations under 21 CFR Part 820 is a regulatory liability — regardless of how competitive its unit pricing is.
Critical FDA requirements that directly affect EMS operations:
- 21 CFR 820.50 — Purchasing controls: EMS providers must evaluate and select suppliers based on their ability to meet specified requirements; supplier qualification records must be maintained
- 21 CFR 820.65 — Traceability: devices intended to be implanted must have traceability to the specific unit level; EMS must support this requirement
- 21 CFR 820.70 — Production and process controls: processes must be controlled through documented procedures, approved materials, and validated equipment
- 21 CFR 820.75 — Process validation: processes whose results cannot be fully verified by subsequent inspection must be validated
- 21 CFR 820.184 — Device History Record: DHR must be maintained for each batch or unit of finished device
For OEMs exporting to Europe, EU MDR (Medical Device Regulation 2017/745) imposes equivalent or stricter requirements — particularly for Class IIa, IIb, and III devices. A medical device EMS provider operating under ISO 13485 provides the quality system foundation that supports both FDA and EU MDR compliance.
>>>Read mor: what is ISO 13485
ISO 13485: The Foundation of Medical Device EMS

ISO 13485 is not a marketing credential — it is the operational framework that determines whether a medical device EMS provider can actually support a regulated medical device program. Understanding what ISO 13485 requires in an EMS context is essential for OEMs evaluating potential partners.
What ISO 13485 Actually Requires From an EMS Provider
ISO 13485:2016 represents the gold standard for quality management systems in medical device manufacturing. Unlike general manufacturing certifications, this standard specifically addresses the unique requirements of producing medical devices. EMS companies with ISO 13485 certification demonstrate their capability to consistently meet both customer requirements and applicable regulatory standards.
This certification is not merely paperwork — it reflects comprehensive quality management systems covering every aspect of production, from design controls and risk management to production validation and post-market surveillance.
Six core requirements of ISO 13485 in an EMS context:
- Document control (Clause 4.2): SOPs, work instructions, and quality records must be controlled, version-managed, and accessible at point of use — not stored in a filing cabinet
- Management responsibility (Clause 5): Quality policy, objectives, and management review must be documented; quality performance must be reviewed at defined intervals
- Resource management (Clause 6): Operator training records, equipment calibration certificates, and facility environmental controls must be maintained and current
- Product realization (Clause 7): Planning, purchasing, production, and process validation must follow documented procedures with defined acceptance criteria
- Measurement, analysis, improvement (Clause 8): Internal audits, nonconforming product control, CAPA, and trend analysis must be active and effective
- Risk management: ISO 14971 integration — risk-based approach to manufacturing decisions, not just quality decisions
| ISO 13485 Clause | EMS Activity | Documentation Required |
|---|---|---|
| 4.2 — Document Control | SOP management, work instructions | Document register, revision history |
| 6.2 — Human Resources | Operator qualification, training | Training records, competency matrix |
| 7.4 — Purchasing | Supplier qualification, incoming inspection | ASL, supplier audits, CoC |
| 7.5 — Production | Process validation, in-process control | IQ/OQ/PQ, batch records |
| 7.6 — Control of Equipment | Calibration, maintenance | Calibration certificates, PM records |
| 8.3 — Nonconforming Product | NCR process, disposition | NCR log, CAPA records |
| 8.5 — Improvement | CAPA, internal audit | CAPA log, audit reports |
ISO 13485 vs. ISO 9001: Why the Difference Matters for Medical OEMs
ISO 9001 is a quality management standard for general manufacturing. ISO 13485 is medical-specific — and the differences are not cosmetic.
Key differences that matter in EMS selection:
- Risk management: ISO 13485 requires a risk-based approach throughout the QMS, integrated with ISO 14971. ISO 9001 does not mandate risk management at this level.
- Validation: ISO 13485 requires validation of software used in production and quality systems. ISO 9001 does not.
- Regulatory compliance: ISO 13485 explicitly requires the QMS to incorporate applicable regulatory requirements. ISO 9001 does not.
- Change control: ISO 13485 requires formal change control with documented impact assessment. ISO 9001 recommends it.
- Record retention: ISO 13485 requires records to be retained for the lifetime of the device or a minimum of two years from release — whichever is longer. ISO 9001 has no medical-specific retention requirements.
Red flag: An EMS provider that claims ISO 9001 is “equivalent” to ISO 13485 for medical device manufacturing does not understand the regulatory landscape. This is a disqualifying response.
How to Verify an EMS Provider’s ISO 13485 Certification
Certification scope is the most commonly overlooked verification step. An ISO 13485 certificate that covers “management services” or “distribution” but not “electronics assembly” does not qualify the EMS provider for medical device manufacturing.
Verification checklist:
- Certificate number and issuing certification body (must be an accredited CB — BSI, SGS, TÜV, Bureau Veritas)
- Scope statement explicitly includes PCBA / electronics assembly for medical devices
- Certificate expiry date and current surveillance audit status
- Site-specific certification — not just corporate-level
- No open major nonconformities from last surveillance audit
Read more: SHDC certifications
Full Traceability in Medical Device Manufacturing
Traceability is where the difference between a medical device EMS provider and a commercial EMS becomes most visible in day-to-day operations. It is also where most EMS providers that claim medical capability fall short.
What “Full Traceability” Means in Medical Electronics Manufacturing
Full traceability in medical device manufacturing is the ability to reconstruct the complete history of a specific unit — from the lot number of every component placed on the board, through every production step and operator, to every inspection result — at any point during the product’s lifetime.
This capability is not just a quality best practice. It is a regulatory requirement under FDA 21 CFR Part 820 and ISO 13485 Clause 7.5.9. When a field failure occurs or a component lot is recalled by a supplier, the OEM must be able to identify — within hours, not weeks — exactly which finished devices contain the affected component. Without full traceability, that identification is impossible.
The four levels of traceability a medical device EMS provider must maintain:
- Component lot traceability: Lot number and date code of every component on every board — linked to the specific unit serial number. This is the foundation of recall response capability.
- Process traceability: Operator ID, machine ID, process parameters, and timestamp for every production step — placement, reflow, inspection, test, conformal coating.
- Inspection traceability: AOI, X-ray, ICT, and FCT results linked to the specific unit serial number — not just a batch pass/fail record.
- Environmental traceability: Temperature, humidity, and ESD conditions during production — critical for moisture-sensitive devices and ESD-sensitive assemblies.
Medical OEMs should expect comprehensive traceability throughout the manufacturing process, including component lot tracking, date code traceability, serialized production records, operator accountability, and inspection and test history. Without strong traceability systems, medical manufacturing risk increases significantly.
Traceability Systems: Manual vs. Digital vs. Integrated MES
| Traceability Type | Data Capture | Recall Response Time | FDA Audit Ready? |
|---|---|---|---|
| Paper-based (manual) | Manual entry | Days–weeks | High risk |
| Digital (standalone software) | Semi-automated | Hours–days | Partial |
| Integrated MES/QMS | Automated at source | Minutes–hours | Yes |
Paper-based traceability is not acceptable for medical device manufacturing — it is error-prone, not searchable, and cannot support the response time required for an FDA recall. An integrated MES/QMS system that captures data automatically at the source — machine, barcode scanner, inspection system — and links it to the unit serial number in real time is the only approach that delivers genuine FDA audit readiness.
Complete traceability forms the backbone of medical device manufacturing quality systems. Full-service contract manufacturers maintain detailed records tracking every component, process step, and operator involved in production. This traceability enables rapid response to any quality issues and supports regulatory requirements for Device History Records.
DHR (Device History Record): What It Must Contain
The Device History Record is a legal document under FDA 21 CFR 820.184. Every finished medical device must have a DHR that demonstrates it was manufactured in accordance with the Device Master Record (DMR). The medical device EMS provider is responsible for generating and maintaining the DHR for every unit produced.
Minimum DHR contents:
- Date of manufacture
- Quantity manufactured and quantity released for distribution
- Primary identification label and labeling used for each production unit
- Equipment identification — serial/lot numbers of critical production equipment
- Component lot numbers and CoC references for all components
- Inspection and test records — AOI, X-ray, ICT, FCT results per unit
- Nonconformance records and disposition for any units with quality events
- Operator and inspector identification for each production step
Internal links: PCBA testing, in-circuit testing ICT, automated optical inspection AOI
FDA-Ready Processes: What This Actually Means in Production
“FDA-ready” is one of the most overused phrases in medical electronics marketing. Here is what it actually means — in terms of specific processes, documentation, and operational discipline — when applied to a genuine medical device EMS provider.
Process Validation: IQ, OQ, PQ in Medical Electronics Manufacturing
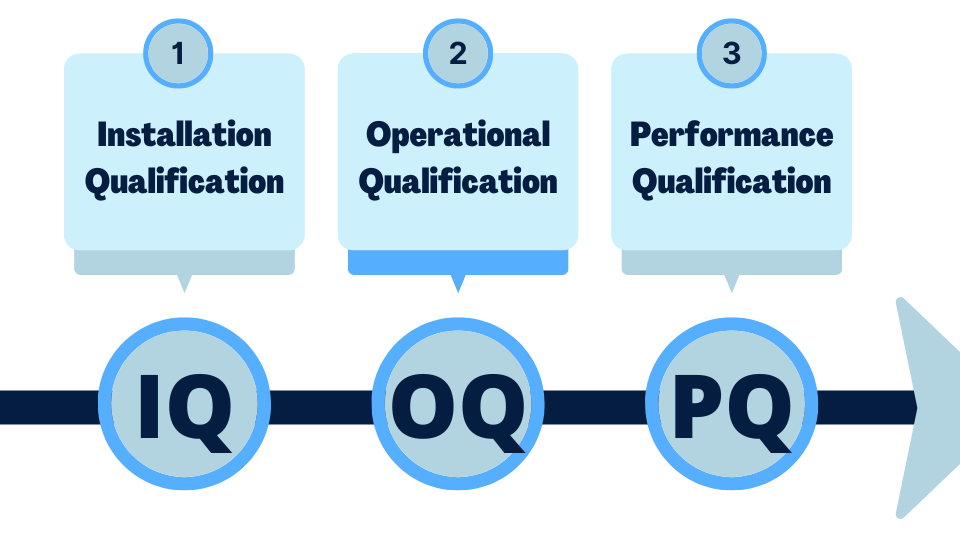
FDA 21 CFR 820.75 requires validation of processes whose results cannot be fully verified by subsequent inspection. In medical electronics manufacturing, this includes reflow soldering, wave soldering, conformal coating, cleaning, and automated inspection.
The three-stage validation framework:
- IQ (Installation Qualification): Confirms that equipment is installed correctly, meets manufacturer specifications, and is operating in the intended environment. Documentation includes equipment specifications, installation records, and calibration certificates.
- OQ (Operational Qualification): Confirms that equipment operates within defined parameters across its full operating range. For a reflow oven, this means validating that the temperature profile meets specification at all positions across the conveyor width, at all production speeds.
- PQ (Performance Qualification): Confirms that the process consistently produces product meeting specification under actual production conditions — using production materials, production operators, and production volumes. This is the stage that demonstrates process capability (Cpk) and establishes the validated operating window.
Critical processes requiring validation in medical PCBA:
| Process | Validation Requirement | Key Parameters |
|---|---|---|
| Reflow soldering | OQ + PQ | Temperature profile, conveyor speed, atmosphere |
| Wave/selective soldering | OQ + PQ | Solder temperature, contact time, flux application |
| Conformal coating | OQ + PQ | Coverage, thickness, cure parameters |
| Cleaning (if applicable) | OQ + PQ | Chemistry, temperature, exposure time, cleanliness verification |
| AOI inspection | Sensitivity validation | Defect detection rate, false call rate |
Change Control: The Most Underestimated FDA Risk in EMS
Change control is where more medical device EMS providers fail FDA audits than any other single area. The reason is simple: in commercial manufacturing, substituting an equivalent component or adjusting a process parameter is routine and informal. In medical device manufacturing, it is a regulated activity that requires documentation, impact assessment, and OEM notification before implementation.
Four categories of changes that must trigger formal change control in a medical device EMS provider:
- Component substitution — including “equivalent” alternate parts. A component that is electrically equivalent may have different physical dimensions, different moisture sensitivity, or different reliability characteristics. Every substitution requires documented impact assessment.
- Process parameter changes — reflow profile adjustment, stencil aperture modification, placement offset correction. Even changes made to improve yield must go through change control.
- Supplier changes — new component manufacturer or distributor, even for the same part number. Supplier changes require incoming inspection validation and may require process re-validation.
- Equipment changes — new production equipment, major maintenance, or repair that could affect process capability.
Change control process flow for a medical device EMS provider:
Change Request → Impact Assessment → OEM Notification & Approval → Validation (if required) → Implementation → Documentation Update → Effectiveness Verification
The most dangerous failure mode: an EMS partner that substitutes a component without notifying the OEM. This is a direct violation of FDA 21 CFR 820.50 and ISO 13485 Clause 7.4 — and it is a potential recall trigger if the substituted component affects device performance or safety.
CAPA System: Corrective and Preventive Action in Medical EMS
CAPA (Corrective and Preventive Action) is the cornerstone of both FDA QSR and ISO 13485. A medical device EMS provider without an effective, documented CAPA system is not FDA-ready — regardless of what their marketing materials claim.
The critical distinction that separates genuine CAPA from superficial quality activity:
- Correction: Fixing the specific nonconforming unit or batch — rework, scrap, or return to supplier. This is the minimum response.
- Corrective Action: Eliminating the root cause of the nonconformance to prevent recurrence. Requires root cause analysis (5-Why, Fishbone, FMEA) — not just symptom correction.
- Preventive Action: Identifying and eliminating potential causes of nonconformance before they occur — based on trend analysis, risk assessment, and process monitoring data.
CAPA system requirements for a medical device EMS provider:
- Root cause analysis methodology documented and consistently applied
- Effectiveness verification — confirming the corrective action actually prevented recurrence
- Trend analysis across CAPA records — identifying systemic issues before they become recurring problems
- Management review of CAPA metrics at defined intervals
- CAPA records maintained and available for FDA audit
Supplier Qualification: The Approved Supplier List (ASL)
FDA 21 CFR 820.50 requires that every medical device EMS provider evaluate and select suppliers based on their ability to meet specified requirements. The Approved Supplier List (ASL) is the documented output of this requirement — and it is one of the first documents an FDA investigator will request during an audit.
ASL requirements for a medical device EMS provider:
- Initial qualification criteria: quality system certification, financial stability, technical capability, delivery performance history
- Ongoing monitoring: delivery performance metrics, incoming inspection results, periodic re-audit schedule
- Component CoC requirements: Certificate of Conformance required for every lot of every critical component
- Counterfeit component prevention: authorized distributor sourcing required for medical applications; broker market components are not acceptable without extensive incoming inspection
When an EMS partner has a robust ASL, the OEM inherits that supplier qualification — reducing the OEM’s own regulatory burden and accelerating the supplier approval process for new programs.
DFM for Medical Device Electronics: Design Decisions That Affect Compliance
Manufacturing partners should contribute engineering expertise early in the process. DFM for electronics manufacturing collaboration helps medical OEMs improve manufacturability, reduce production risk, improve yield and reliability, and accelerate product launches. Strong engineering collaboration becomes especially important during New Product Introduction and scaling phases.
Medical-Specific DFM Considerations
DFM for medical electronics goes beyond standard commercial DFM — it must address traceability infrastructure, IPC Class 3 design rules, and regulatory documentation requirements simultaneously.
Key medical-specific DFM requirements:
- Traceability by design: PCB layout must include dedicated space for serial number marking — laser-etched or 2D barcode. If the layout does not accommodate a scannable serial number, the traceability system cannot function. This is a design decision, not a production decision, and it must be made at DFM stage.
- IPC Class 3 design rules: tighter minimum annular rings, wider trace/space for reliability, thermal relief design for through-hole components on internal power planes — all must be verified at DFM review before the first build
- Conformal coating compatibility: component placement must allow coating nozzle access; connectors must have masking provisions; keep-out zones around coating-sensitive components must be defined
- Test point accessibility: ICT coverage ≥95% is the target for medical electronics — DFT review must happen at design stage, not after the ICT fixture is already being built
- Component height and clearance: conformal coating nozzle access and X-ray inspection angle requirements impose specific keep-out zones that must be designed in, not retrofitted
Component Selection for Medical Reliability
Medical devices have product lifecycles of 7–15 years. Component selection decisions made at design stage have regulatory and supply chain implications that extend across the entire device lifetime.
Critical component selection requirements for medical electronics:
- Long lifecycle components: components approaching EOL at design stage will require a design change — with associated regulatory re-evaluation — before the device reaches end of market life. A medical device EMS provider with active EOL monitoring can identify these risks at BOM review stage.
- Moisture sensitivity management: MSL ratings and baking requirements for all ICs must be documented and enforced in production. Moisture-damaged components that pass incoming inspection may fail in the field — a medical device reliability risk that is entirely preventable.
- Counterfeit component prevention: authorized distributor sourcing is mandatory for medical applications. Components from broker markets require extensive incoming inspection including decapsulation and electrical characterization — costs that make broker sourcing economically unviable for most medical programs.
- AEC-Q100/Q200 qualified components: for high-reliability medical applications, components qualified to automotive reliability standards provide additional assurance of long-term performance
NPI Process for Medical Devices: From EVT to FDA Submission
Medical NPI has an additional layer of complexity that commercial NPI does not: design freeze. Every change made after design freeze requires regulatory re-evaluation — and if the device is a Class II or Class III device, it may require a new 510(k) submission or PMA supplement.
The NPI process for medical devices must be structured to support design freeze at the right point — after all manufacturing process issues are resolved, but before regulatory submission:
| NPI Stage | Medical DFM Activity | Regulatory Milestone |
|---|---|---|
| EVT | Full DFM + DFT review; medical-specific checklist | Design input review |
| DVT | IQ/OQ initiation; IPC Class 3 inspection baseline | Design verification |
| PVT | PQ completion; DHR template validation; full traceability test | Design validation; design freeze |
| MP | Control Plan enforcement; ongoing process monitoring | FDA submission support |
Internal link: prototype PCBA manufacturing
Why Vietnam for Medical Device EMS? The Strategic Case for U.S. OEMs

The strategic case for sourcing medical device EMS from Vietnam in 2026 is built on four converging factors: tariff advantage, ISO 13485 availability, engineering capability, and geopolitical stability.
Vietnam’s Medical Electronics Manufacturing Ecosystem
Vietnam has emerged as a serious medical electronics manufacturing destination over the past five years — driven by investment in quality infrastructure, IPC certification programs, and the migration of U.S. OEM programs away from China.
Key advantages for U.S. OEMs sourcing medical device EMS from Vietnam:
- Tariff advantage: U.S. import tariffs on Vietnam electronics are significantly lower than the 145% tariffs imposed on Chinese electronics manufacturing in 2025. For medical devices with high unit values, this tariff differential directly impacts product economics and market competitiveness.
- ISO 13485 availability: A growing number of Vietnam EMS providers have invested in ISO 13485 certification — making medical-grade manufacturing available at Vietnam’s cost structure
- Engineering talent: Vietnam’s electronics engineering workforce has grown substantially, with increasing numbers of IPC-certified technicians and process engineers trained in medical manufacturing requirements
- ASEAN component access: Vietnam’s position within the ASEAN manufacturing ecosystem provides access to component supply chains that reduce lead times compared to purely domestic sourcing
Read more: why Vietnam is the top choice for electronics manufacturing in 2026, 10 reasons to choose contract electronics manufacturing in Vietnam
Vietnam vs. China for Medical Device EMS
| Factor | China EMS | Vietnam EMS (SHDC) |
|---|---|---|
| U.S. import tariff | 145% (2025+) | ~10–20% |
| ISO 13485 availability | Widespread | Growing — SHDC certified |
| IP protection risk | High | Lower, improving legal framework |
| FDA audit readiness | Variable | Increasing U.S. OEM engagement |
| Component ecosystem | Largest globally | ASEAN access + direct import |
| Geopolitical risk | High | Low |
| English-capable engineering | Variable | Available at SHDC |
| Regulatory documentation | Variable quality | Structured, audit-ready at SHDC |
Internal links: China alternative electronics manufacturing, offshore PCB assembly
Transfer Products: Moving Medical Device Manufacturing to Vietnam
Transferring a medical device program from an existing manufacturer — whether China-based or domestic U.S. — to a Vietnam medical device EMS provider requires a structured transfer validation process. Process assumptions from the previous manufacturer do not automatically transfer, and yield regression at transfer is a known risk without proper validation.
Transfer validation requirements for medical device programs:
- Process equivalency study: demonstrating that the new facility’s processes produce statistically equivalent results to the previous facility
- First Article Inspection (FAI) with complete DHR: first units built at the new facility must be fully documented and inspected against the Device Master Record
- Statistical process capability (Cpk) comparison: process capability at the new facility must meet or exceed the validated capability at the previous facility
- FDA notification: depending on device class and the nature of the manufacturing change, FDA notification or submission may be required before commercial production begins at the new facility
Internal links: offshore manufacturing risks, electronics supplier due diligence, electronics factory audit checklist
SHDC as Your Medical Device EMS Provider in Vietnam

SHDC Electronics Company is a full-service medical device EMS provider located at the Vietnam Singapore Industrial Park — Hai Duong (VSIP Hai Duong), 40km from Hanoi and 55km from Haiphong Port. With a 2,600 m² facility, 150 employees, and a dedicated Engineering Department comprising Product Technical and Technical Factory teams, SHDC operates the quality infrastructure that medical device manufacturing requires — not as an add-on service, but as the foundation of every program.
SHDC’s Medical-Grade Infrastructure
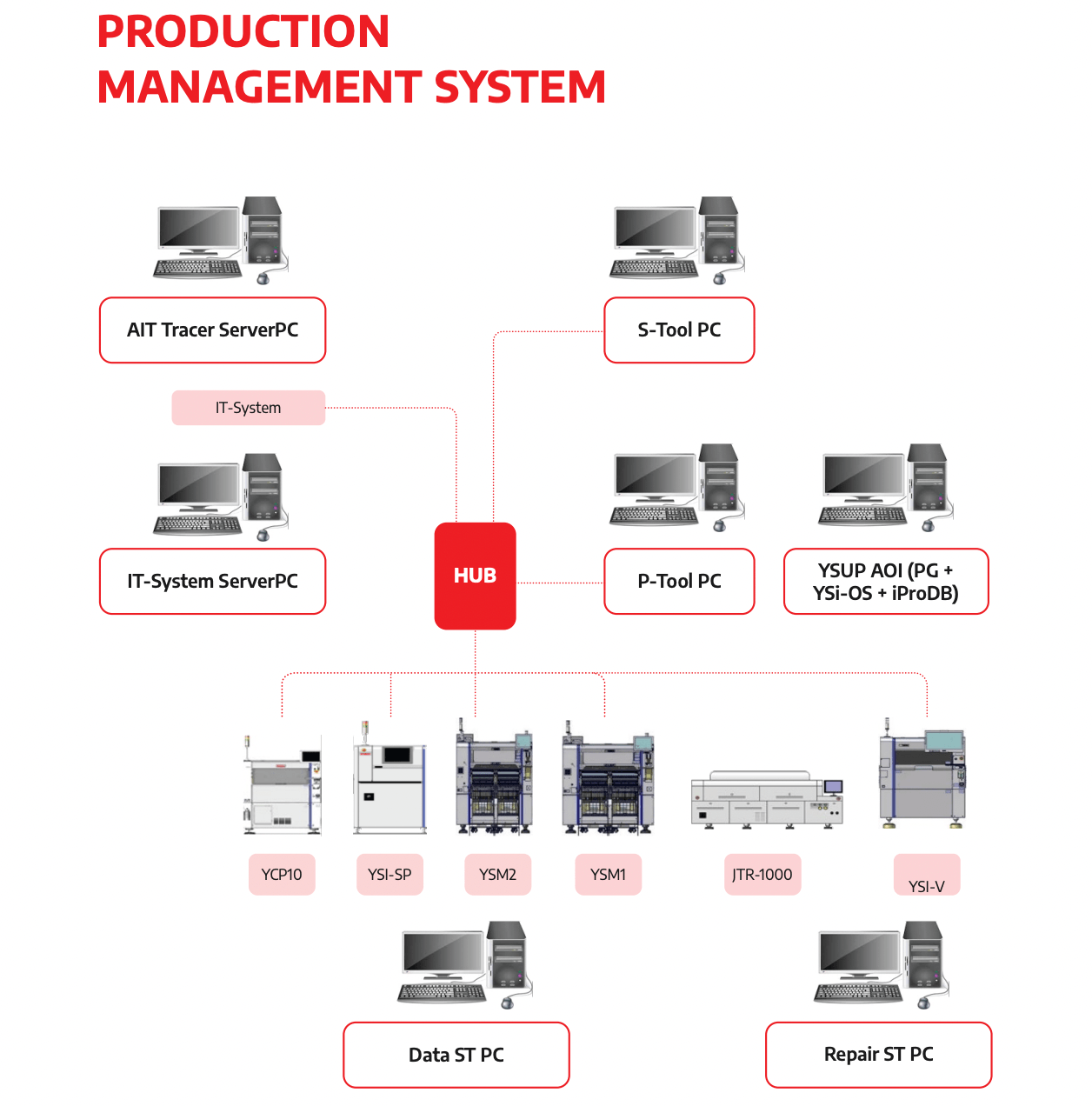
Integrated ERP/MES/QMS with AIT Tracer SHDC operates an integrated ERP/PLM/SCM/MES/QMS digital infrastructure. The AIT Tracer system provides board-level traceability from the first prototype build — component lot numbers, process parameters, operator IDs, and inspection results are captured automatically and linked to unit serial numbers in real time. This is the integrated MES/QMS traceability architecture that supports FDA audit readiness and recall response capability. According to SHDC’s production management data, this integration reduces decision-making time by 30% and reduces defective inventory by 30%.
IPC Class 3 Capability IPC-A-610 certified inspectors and J-STD-001 certified operators are active on SHDC’s production floor. Class 3 inspection criteria are applied from the first prototype build — not introduced at pilot or production stage. This means the inspection baseline established during NPI is the same baseline used in mass production.
Yamaha Smart Factory — 3D SPI and 3D AOI The Yamaha YSI-SP 3D Solder Paste Inspection system validates stencil design before the first reflow cycle. The Yamaha YSI-V 3D AOI system is active from the first prototype build, with inspection data linked to unit serial numbers automatically. For medical electronics, this means every unit has a documented, traceable inspection record from its first production step.
Precision Placement Capability
| Equipment | Placement Accuracy | Speed | Component Range |
|---|---|---|---|
| Yamaha YSM20R | ±0.03mm (Cpk ≥ 1.00) | 95,000 CPH | 0402 to 32×32mm |
| Yamaha YSM10 | ±0.035mm | 46,000 CPH | 03015 to 55×100mm |
ICT and Functional Testing The Kyoritsu ICT F-2000 Plus provides parametric in-circuit test capability. Dedicated FCT stations, aging test equipment, high-voltage test, and AV test infrastructure support comprehensive functional validation. ICT fixture development is initiated at DVT stage — ensuring test infrastructure is ready when the pilot run completes, not weeks after.
SHDC’s Medical Device Manufacturing Track Record
SHDC has applied medical-grade process discipline across a range of demanding product categories:

Thaco Automotive PCBA: Automotive PCBA Vietnam — PPAP-level documentation, the most rigorous documentation standard in manufacturing, directly applicable to medical device DHR requirements

GaN Power Electronics (65W–150W): GaN charger manufacturing in Vietnam — high-density, thermally demanding designs with IPC Class 3 processes and full traceability
Industrial Electronics: Industrial electronics manufacturing in Vietnam — complex mixed-technology assemblies with controlled process documentation
What to Expect From SHDC’s Medical Device EMS Onboarding
SHDC’s medical device program onboarding follows a structured sequence designed to establish regulatory defensibility from day one:
- NDA execution — IP protection before any design data is shared
- Design package review — DFM for electronics manufacturing analysis with medical-specific checklist: traceability marking space, IPC Class 3 design rules, conformal coating compatibility, ICT coverage ≥95%
- Quality plan development — Control Plan, PFMEA, and inspection criteria aligned to IPC Class 3 and ISO 13485 requirements
- Supplier qualification — component sourcing from SHDC’s ASL; new components qualified against medical requirements including authorized distributor verification
- Process validation — IQ/OQ/PQ documentation developed before first production build; reflow profile, placement parameters, and inspection sensitivity validated
- Prototype build + DHR — first unit built with complete DHR template validated against FDA 21 CFR 820.184 requirements
- FDA audit readiness review — documentation package reviewed against 21 CFR Part 820 requirements before pilot run
Phase 2 Expansion — March 2027

SHDC’s new facility at Lai Cach Industrial Park will add 10 SMT lines, 8 DIP lines, 10 assembly lines, and full inline ICT/FCT/AOI 3D capability — scaling capacity to 50M units/year. Medical device programs started today will have a clear, validated path to high-volume production within the same manufacturing ecosystem — with no EMS transfer risk, no process re-validation, and no regulatory re-qualification at ramp.
Frequently Asked Questions
What is a medical device EMS provider?
A medical device EMS provider is a contract electronics manufacturer that operates under a quality management system specifically designed for medical device production — typically ISO 13485 certified — with full component traceability, process validation, formal change control, and FDA-ready documentation systems. Unlike commercial EMS providers, a medical device EMS provider builds to IPC Class 3 standards and maintains Device History Records for every unit produced.
What certifications should a medical device EMS provider have?
The minimum certification requirement is ISO 13485 with a scope statement that explicitly covers electronics assembly for medical devices. Additional relevant certifications include IPC-A-610 operator certification (Class 3), J-STD-001 soldering certification, and ISO 9001. For programs with automotive-equivalent documentation requirements, IATF 16949 experience is also relevant.
Is ISO 13485 required for medical device contract manufacturing?
ISO 13485 is not legally mandated by FDA regulation — but it is the recognized quality management standard for medical device manufacturing and is required by EU MDR for manufacturers supplying the European market. More practically, most medical device OEMs require their medical device EMS provider to be ISO 13485 certified as a condition of supplier qualification.
What is the difference between ISO 13485 and ISO 9001 for medical devices?
ISO 13485 is medical-specific and requires risk management (ISO 14971 integration), process validation, formal change control, and regulatory compliance as core QMS elements. ISO 9001 is a general quality standard that does not address these medical-specific requirements. An EMS provider with only ISO 9001 is not qualified for medical device manufacturing.
What does full traceability mean in medical device manufacturing?
Full traceability means the ability to reconstruct the complete history of a specific unit — component lot numbers, process parameters, operator IDs, inspection results, and environmental conditions — linked to a specific unit serial number, at any point during the product’s lifetime. This capability supports FDA recall response, failure analysis, and regulatory audit.
What is a Device History Record (DHR) and who is responsible for it?
The DHR is a legal document under FDA 21 CFR 820.184 that records how a specific device was manufactured. It must contain the date of manufacture, quantity produced, component lot numbers, equipment identification, inspection and test records, and any nonconformance records. The medical device EMS provider is responsible for generating and maintaining the DHR for every unit produced.
What FDA regulations apply to medical device EMS providers?
Key FDA regulations applicable to medical device EMS providers include: 21 CFR Part 820 (Quality System Regulation) — particularly 820.50 (purchasing controls), 820.65 (traceability), 820.70 (production controls), 820.75 (process validation), and 820.184 (Device History Record). EMS providers are subject to FDA inspection as suppliers to medical device manufacturers.
How do I qualify a new EMS provider for medical device manufacturing?
Qualification of a new medical device EMS provider should include: ISO 13485 certificate verification (scope, issuing body, expiry), on-site quality system audit, process capability assessment, traceability system demonstration, review of CAPA records and internal audit history, and a qualification build with full DHR review. See electronics factory audit checklist and electronics supplier due diligence.
Can a Vietnam EMS provider manufacture FDA-regulated medical devices?
Yes. FDA regulations apply to the device and its OEM — not to the geographic location of the contract manufacturer. A Vietnam medical device EMS provider operating under ISO 13485 with FDA-ready process documentation can manufacture FDA-regulated medical devices. FDA may inspect the contract manufacturer’s facility as part of an OEM audit. SHDC’s quality infrastructure is designed to support FDA audit readiness.
What is IPC Class 3 and why does it matter for medical electronics?
IPC Class 3 is the highest reliability level defined in IPC-A-610 — designed for electronics where continued performance is critical and equipment downtime cannot be tolerated. Class 3 imposes tighter solder joint acceptance criteria, 100% inspection requirements, and mandatory X-ray inspection for hidden solder joints. Medical electronics must be built to IPC Class 3 to meet the reliability requirements of a regulated medical device.
How does change control work in medical device EMS?
Every change to component, process, or supplier at a medical device EMS provider must go through formal change control: change request, impact assessment, OEM notification and approval, validation (if required), implementation, and documentation update. Component substitutions made without OEM notification violate FDA 21 CFR 820.50 and ISO 13485 Clause 7.4 — and may constitute a recall trigger.
What is CAPA and why is it important for medical device manufacturing?
CAPA (Corrective and Preventive Action) is a core requirement of both FDA QSR and ISO 13485. It requires root cause analysis of nonconformances (not just symptom correction), implementation of corrective actions that prevent recurrence, and preventive actions that eliminate potential failure modes before they occur. An effective CAPA system is the primary mechanism by which a medical device EMS provider demonstrates continuous quality improvement to FDA auditors.
Conclusion: Medical Device EMS Is Where Regulatory Risk Is Won or Lost
The medical device EMS provider you choose is not a vendor — it is a co-owner of your regulatory risk. Their ISO 13485 certification, traceability system, process validation records, change control discipline, and CAPA effectiveness will appear in your FDA audit, your DHR, and your recall response capability. Choosing a partner based on unit price without verifying these capabilities is the most expensive procurement decision a medical device OEM can make.
For U.S. OEMs evaluating Asia-Pacific manufacturing options in 2026, Vietnam offers a compelling combination: ISO 13485-certified EMS capability, significantly lower tariff exposure versus China, and the engineering infrastructure to support FDA-ready medical device production from prototype through mass production. The regulatory trajectory — particularly the increasing FDA scrutiny of offshore contract manufacturers — makes the quality system of your medical device EMS provider more important, not less, as programs scale.
The right medical device EMS provider doesn’t just build your boards — they build the documentation, traceability, and process validation infrastructure that makes your device defensible in front of an FDA investigator, a notified body auditor, and a plaintiff’s attorney.
